HIVprotI
HIV Protein Inhibitor Prediction
Algorithm
HIV protein inhibitors
The human immunodeficiency virus (HIV) causes acquired immunodeficiency syndrome (AIDS) in which gradual breakdown of the immune system allows critical opportunistic diseases to flourish. Different HIV enzymes are needed for the development of the retrovirus including reverse transcriptase (RT), protease (PR) and integrase (IN). RT creates complementary DNA from an RNA template which can integrate into the host genome. RT inhibitors are widely used as antiretroviral drugs. PR slices the newly synthesized polyproteins at the pertinent positions to form the mature protein apparatus. PR is a major drug-target for treatment of HIV. The IN enzyme enables the virus to integrate its genetic material into the DNA of the host cell for a long-term infection. Compounds that inhibit the IN enzyme have demonstrated potent anti-HIV activity. Blocking the action of these indispensable HIV proteins is the subject of considerable pharmaceutical research.
Datasets
We used the data from the ChEMBL (https://www.ebi.ac.uk/chembl/target/browser/). In this project,we selected RT, PR and IN inhibitors(protein specific datasets). The three enzymes were respectively targeted by 2126, 1895, 1240 (IC50) and 563, 518, 186 (percent inhibition) unique compounds in the database. More details can be found here and here.
Chemical Features
To develop QSAR models, we have calculated the different chemical descriptors (1D, 2D and 3D) and fingerprints by using PaDEL software (http://padel.nus.edu.sg/software/padeldescriptor/). These relevant chemical descriptors were extracted using attribute selection algorithms viz. 'RemoveUseless' filter followed by ClassifierSubsetEval (attribute evaluator) with BestFirst (search method) module available in Weka package.
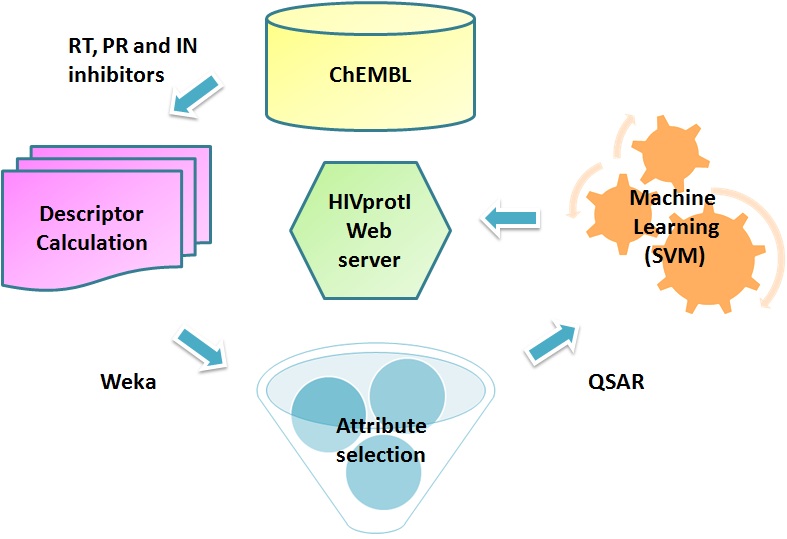
QSAR Models
We have developed separate QSAR models for each of the three HIV proteins (RT, PR and IN) by using SMOreg algorithm available in Weka machine learning package. SMOreg implements the support vector machine in regression mode. Selected molecular descriptors calculated by the PaDEL were used as input.
Cross Validation
All models were evaluated using ten-fold cross validation technique as well as on independent test datasets. In order to evaluate performance of our models, we used Pearson’s correlation coefficient (R).
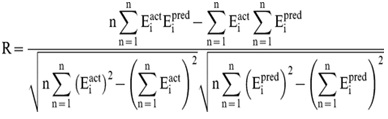
Where n is the size of test set, Eipred and Eiact is the predicted and actual efficacy respectively.

